We are interested in developing computational technologies to understand the functions of coding and non-coding elements, especially in the context of human physiology and disease. We are focusing on the following areas:
Area 1: Developing innovative methods for genome engineering and functional genetic screens
Example 1: modeling gene editors
- DeepCas13 (for Cas13)
- BEEP (for base editing)
- CRISPR-FOCUS
- SSC
Our most recent finding (published in Nature Biomedical Engineering 2023; in collaboration with Teng Fei lab at Northeastern University) reveals a surprising off-target mechanism of Cas13, which can intrinsically target host RNA in mammalian cells through previously unappreciated mechanisms. This intrinsic RNA targeting mechanism limits the application of Cas13 using viral vectors as delivery methods.
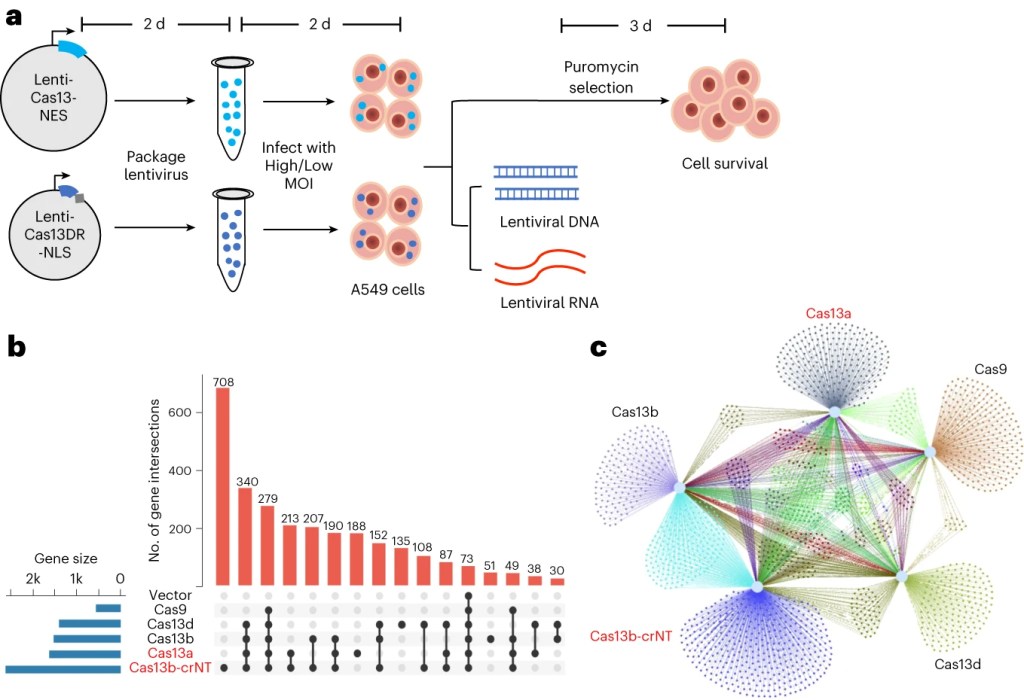
Figure 1. Intrinsic RNA targeting of Cas13. See Research Briefing.
Example 2: pooled and single-cell CRISPR screens
We developed algorithms for the modeling and processing of pooled CRISPR screens, which are widely used by the community:
In addition, we developed algorithms for modeling single-cell CRISPR screens:
Databases for large-scale genetic screens spanning multiple phenotypes:
And so on. These algorithms became popular in the field: MAGeCK reaches over 1600 citations and over 120,000 software downloads. These softwares enabled researchers to identify interesting hits from screens, and to perform joint analysis from multiple screening experiments:
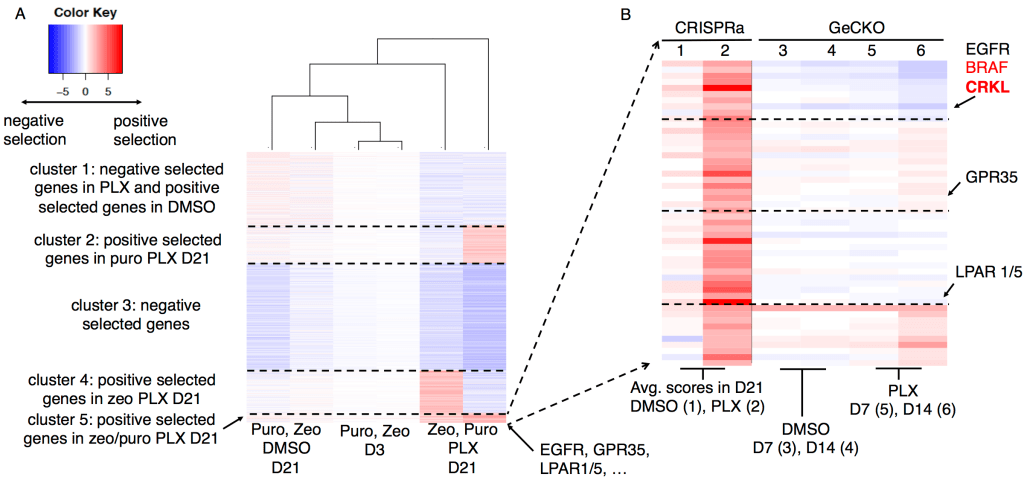
Figure 2: Analyzing gene functions using MAGeCK-VISPR in a single experiment (left) and two experiments (right)
Area 2: Modeling human genome functions using gene editing, AI, and single-cell genomics
Using the computational frameworks we developed, we collaborated with experimental and clinical scientists around to world to study human genome functions and their associations with human diseases.
Example 1: understanding gene perturbations at a single cell level
CRISPR/Cas9-based functional screening coupled with single-cell RNA-seq (“single-cell CRISPR screening”, or Perturb-seq) is an exciting new technology that combines genome engineering with single-cell sequencing. It’s particularly helpful to understand gene regulatory networks and enhancer-gene regulations on a large scale. We propose scMAGeCK, a computational framework to systematically identify genes and non-coding elements associated with multiple expression-based phenotypes in single-cell CRISPR screening. scMAGeCK is a novel and effective computational tool for studying genotype-phenotype relationships at the single-cell level. scMAGeCK was published in Genome Biology.
Our most recent work introduces the Perturbation-response score (PS), which models heterogeneity in Perturb-seq. We developed a PS analysis method to unlock the full potential of single-cell perturbation datasets. This method enables an innovative dosage analysis of genetic perturbations and the identification of biological determinants that control heterogeneous responses to perturbations. The paper is published in Nature Cell Biology.
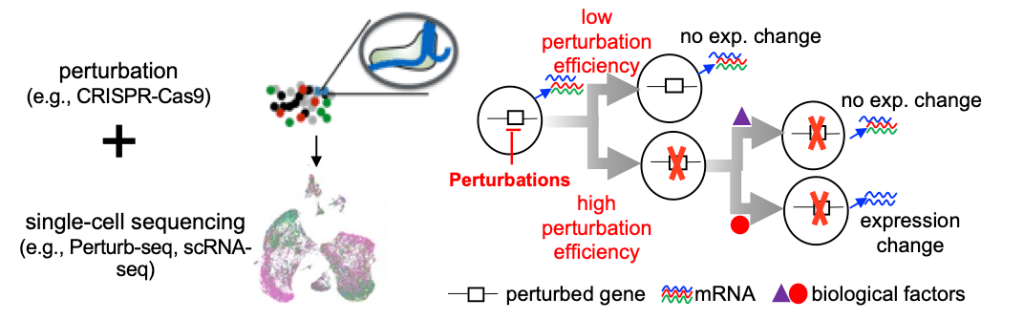
Figure 3. Perturbation-response score (PS) to analyze heterogeneity in single-cell CRISPR screens (Perturb-seq)
Example 2: novel drug target discovery in breast cancer and infectious diseases
Latent HIV infection. Human immunodeficiency virus type 1 (HIV-1) persisting in a latent form in resting CD4+ T cells despite effective antiretroviral therapy (ART) is the major barrier to cure. A promising therapeutic approach known as “shock and kill” seeks to achieve a cure by sequentially reversing latency in infected cells and then killing the productively infected cells. Latency-reversing agents (LRAs) that act through various proposed mechanisms have been identified. To date, however, no single LRA (or LRA combination) has been shown to reduce latent reservoir size in persons living with HIV-1 (PLWH).
We collaborated with Robert Siliciano lab (JHU) to develop an approach to systematically identify LRA combinations to reactivate latent HIV-1 using genome-wide CRISPR screens. We identified several synergistic LRA combinations, including AZD5582, HDAC inhibitors, and the BET inhibitor, JQ1. Moreover, we identified and validated additional synergistic drug candidates for which no drugs had been developed, including CYLD and YPEL5. Our study provides insights into the roles of host factors in HIV-1 reactivation and validates a system for identifying drug combinations to reverse HIV-1 latency. This study was published in Science Translational Medicine 2022.
Breast cancer. Over 70% of breast cancer patients are ER positive, and endocrine therapy has been a standard treatment for these patients for decades. However, most patients with advanced-stage disease will eventually develop resistance to ER inhibition therapies with unknown mechanisms. We collaborated with Myles Brown’s lab (at Dana-Farber Cancer Institute/Harvard Medical School) to study the mechanisms underlying endocrine resistance in breast cancer and potential treatment strategies. By analyzing genome-wide CRISPR knockout screening data, we identified an unusual tumor suppressor, c-src tyrosine kinase (CSK), whose loss accelerated cell growth in the absence of hormones and was associated with high-grade tumors and poorer survival rates in patients.
We also identified genes that are synthetic lethal in CSK loss from screens that can serve as drug targets. The top hit (PAK family kinase) is confirmed as a vulnerable target for endocrine-resistant patients, and the small molecule PAK inhibitor suppresses tumor growth in various confirmation experiments. In other words, we not only found a biomarker that is responsible for breast cancer drug resistance, but also found a potential drug that can be repurposed to treat these patients. The paper was published in PNAS 2018, and a corresponding patent application was submitted.
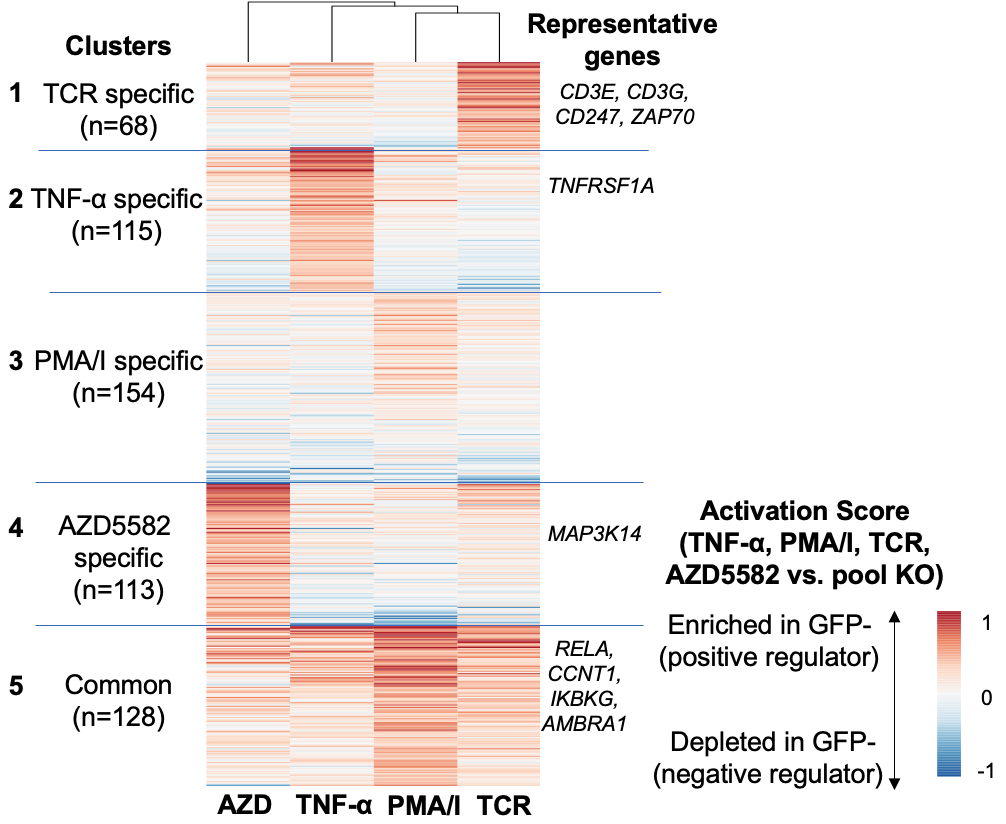
Figure 4. Genome-scale CRISPR screens to find critical genes for HIV latency.
Example 3: Studying functional long non-coding RNAs in cancer
Long non-coding RNAs (lncRNAs) do not translate into protein, but they are important in many aspects (including cancer). In collaboration with Wensheng Wei’s laboratory (Peking University), we developed a novel computational and experimental protocol to screen for lncRNAs using paired gRNAs (pgRNAs). This technology introduces pgRNAs simultaneously into one cell, and is able to efficiently knock out non-coding elements by introducing large genomic deletions. We demonstrated its ability to knock out lncRNAs rapidly and efficiently. The paper was published in Nature Biotechnology.
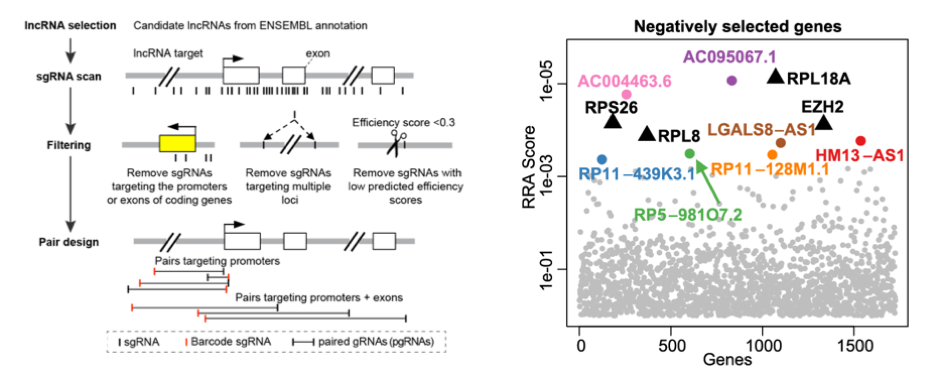
Figure 5. lncRNA screening: designing algorithm (left) and identifying top hits (right) using CRISPR screening.
Example 4: Transcriptome dynamics from RNA-seq and scRNA-seq
RNA-Seq is an exciting technology to study transcriptome via the second generation sequencing. We studied the problem of de novo transcriptome assembly from RNA-Seq reads — reconstructing all possible message RNA compositions simultaneously, without using any information from current gene annotations. We developed a series of influential algorithms for RNA-seq transcriptome assembly and expression analysis: IsoInfer, IsoLasso, CEM and ISP. IsoInfer and IsoLasso were the first algorithms to use combinatorial methods and regularized least squares methods to study assembly problem in RNA-seq.
We are now working on single-cell RNA-seq (scRNA-seq), an exciting new technology to study transcriptome dynamics at the single-cell level.
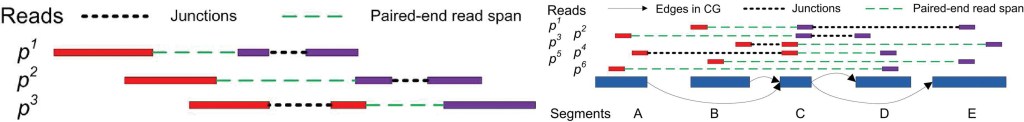
Figure 6. The IsoLasso splicing model
